Application Note Threshold Total DNAアッセイシステムを用いた
最終製品バイオ医薬品中の残留DNAの定量。
アッセイのバリデーションと検出限界の決定
PDF版(英語)
はじめに
FDAは、バイオ医薬品のリリースに使用される全ての品質管理試験について、連邦規則集Title 21に概説されている現行の適正製造規範(cGMP)に従うことを推奨しています。品質管理試験のバリデーションは、精度、正確性、直線性(感度を含むアッセイ応答曲線の特徴)、 検出限界、システム適合性(クロマトグラフィーアッセイにのみ必要)及び特異性に関して、許容できる 性能を提供することを証明しなければなりません1 。最終製品のバイオ医薬品に残存するDNAを定量するために使用される検査(Threshold® Total DNAアッセイ、ハイブリダイゼーションなど)はこのカテゴリーに属し、これらの基準に従ってバリデートされるべきです。
Threshold Total DNA アッセイ2 については直線性と特異性が既に評価されているため、Threshold システムを用いたルーチンの DNA 定量に先立ち評価すべきその他の関連性能基準は、精度、確度、検出限界です。精度は、サンプルに添加したDNAの回収率(スパイク回収率)を評価することで実証できます。精度は、1日以内、1日ごと、オペレーター間、検査室間の性能試験を通じて評価することができます。検出限界は、採用する試料の前処理方法と必要な信頼水準(95%、99.9%など)の関数となります。
このアプリケーションノートでは、定量精度を検証するための1つの手法を提案しています。さらに、実際のバイオ医薬品サンプルおよび緩衝液における残留DNAの定量および検出限界の決定に関する技術も提案しています。本研究では、最も効率的な前処理プロトコールが既に確立されており3、適切なDNA標準物質が選択され、調製され、適格性が確認されていることを前提としています4。
材料と方法
バイオ医薬品(ヒト成長ホルモン[rHGH]、IgGおよびプロテインX)の最終製品(製剤化またはバルク、プレフォーミュレーション)はThresholdユーザーから供給されました。SDS(Bio-Rad電気泳動グレード)の2%(w/v)溶液をZero Calibrator(50 mM Phosphate buffered saline at pH 7.0、Threshold Total DNA assay kitに付属)で調製しました。Proteinase K [Boehringer Mannheim Biochemicals (Indianapolis, IN)] 2 mg/ml溶液もZero Calibratorで調製しました。
この研究では、既知量の添加DNA(スパイク)を含むサンプルを以下の方法で調製した: 1) DNAストック溶液を適切な希釈液(例えば、IgGおよびrhGH試験用のコントロールバッファーまたは1 mg/mlタンパク質溶液)に連続希釈する、または2) 既知量のDNAを含むサンプル(例えば、プロテインX用のコントロールバッファーまたは1.6 mg/mlタンパク質溶液)を個別にスパイクします。全てのサンプルは3回に分けて調製され、滅菌されたスクリューキャップ付きチューブで一晩プロテアーゼ消化されました。DNA熱変性ステップの前のすべてのサンプル取り扱いステップにおいて、無菌技術が観察されました。
Threshold Systemでの試料の前処理およびアッセイに使用する推奨および必要な材料(滅菌ピペットチップやチューブなど)のリストは、Threshold System Operator's Manualに記載されています。Threshold装置および試薬キットはMolecular Devices Corporation(カリフォルニア州サニーベール)から供給されました。タンパク質の消化、正常標準曲線の作成、Threshold Systemおよびソフトウェアの操作については、Threshold System Operator's Manualに概説されているプロトコルに従いました。
結果と考察
Thresholdバイオ分析システムソフトウェアTHSは、0.5mlサンプルあたりのDNA濃度を質量単位で計算します。結果として得られる定量値には、前処理段階で混入した可能性のあるDNAが含まれます。バイオ医薬品1mgあたりのDNA濃度を報告する前に、この外来性DNAを差し引く必要があります。従って、適切な標準物質と対照物質が含まれていれば、試料中に残存するDNAの定量値を手動で計算することができます。定量精度は、未知試料に添加したDNAの回収率を、未知試料と同一の方法で処理したバッファーコントロールに対する相対値で評価することにより評価することができます。
前処理法のバリデーション
定量精度の検証においては、標準曲線の形状と傾きが、前処理法および/または試料成分の影響を(試験法の実験誤差の範囲内で)受けないことを示すことが重要です。前処理方法がDNA回収率に及ぼす影響を判断する一つの方法は、熱変性(一本鎖)DNAから作成した標準曲線(Threshold System Operator's Manualで提案されている「通常の標準曲線」)と、試料が提案されている試料前処理方法で処理された二本鎖DNAから作成した標準曲線を比較することです。図1にそのような比較の例を示します。この場合の前処理法は、55℃でSDSとProteinase Kで一晩処理することです。許容可能なスパイク回収率は通常、適切な対照試料と比較した場合、100±20%(ラボの好みによる)と定義されます。図1に示した比較のための標準曲線の線形回帰分析は、標準曲線の低い直線端だけを調べるので十分です。回帰分析により、緩衝液と通常の標準曲線について以下の式が得られます:
| \( y = 0.84x + 6.3 \) | R² = 0.998 | 緩衝液 |
|---|---|---|
| \( y = 0.82x + 1.2 \) | R² = 0.999 | 正常標準曲線 |
上記のスパイク回収ガイドラインに従えば、処理された各サンプルポイントは対応するコントロールの20~30%以内にあるはずです。したがって、2本の直線はほぼ平行になるはずです。2つの直線の傾きを比較すると、わずか2.5%の差しかないことがわかります。さらに、回帰係数はどちらも0.998より大きいので、この線形分析は有効であり、標準曲線の形状は使用する前処理方法と関連試薬の影響を受けないことを示しています。
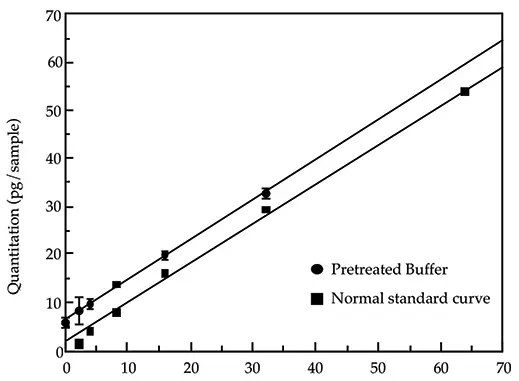
図1: 前処理した緩衝液と通常の標準曲線の比較。前処理した緩衝液の標準曲線ポイントの定量は、通常の標準曲線に基づいて行いました。前処理されたサンプルはバルク溶液として調製され、一晩の消化のために三連に分けられ、翌朝アッセイされました。前処理は、0.1% SDSと100 µg/ml Proteinase Kの存在下、55℃で一晩消化しました。
前処理法およびそれに関連する試薬によって導入される可能性のある外来性DNAの決定は、前処理した緩衝液と通常の標準曲線との間のy切片の差を取ることによって計算することができます。この例では、差は 5.1 pg です。したがって、5.1 pg の DNA がサンプルの前処理中に導入されたことになります。この量は、同じ実験において同様の方法で処理されたバイオ医薬品サンプルの残留 DNA を測定するための手動計算におけるベースラインとして使用する必要があります。
サンプル中の残存DNAの検証と定量
同様の分析を用いて、前処理したバイオ医薬品サンプルを前処理したバッファーコントロールと比較することができます。図2および図3は、最終製品IgGおよびrhGHサンプルのこのような比較を示しています。線形回帰分析によると、回帰係数はすべての行で0.97を超えています。いずれの場合も、緩衝液コントロールとタンパク質サンプルのポイント間の定量値の差はごくわずかです(許容できる統計誤差の範囲内)。したがって、前処理されたサンプルと前処理されたバッファーの回帰直線はほぼ平行です。
また、両タンパク質のy-切片の差は≤1 pgです。これらの事実と簡略化のため、これらのグラフでは、すべての前処理サンプルのデータを表すために1本の線を使用しています。
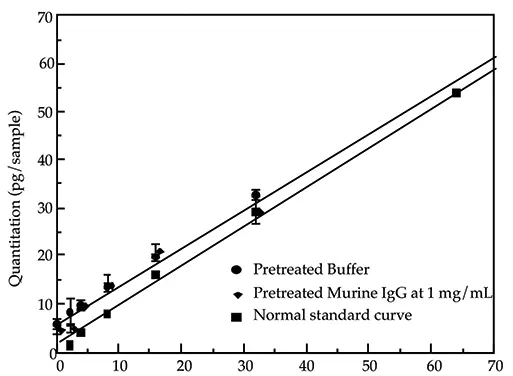
図2: 前処理した緩衝液および前処理した最終製品IgGサンプルと通常の標準曲線との比較。前処理した緩衝液サンプルの定量は、通常の標準曲線に基づいて行いました。前処理サンプルはバルク溶液として調製され、一晩の消化のために三連に分離され、翌朝アッセイされました。前処理は、0.1% SDSと100 µg/ml Proteinase Kの存在下、55℃で一晩消化しました。
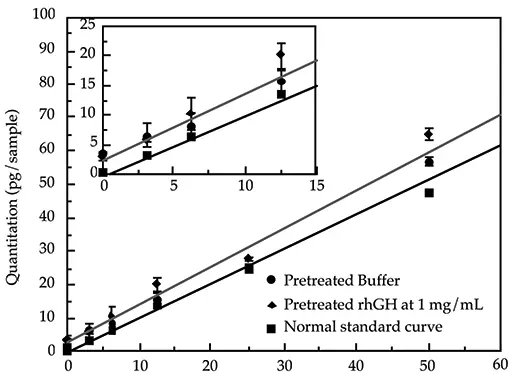
図3: 前処理した緩衝液と前処理した最終産物のrhGHと通常の標準曲線との比較。前処理した緩衝液サンプルの定量は、通常の標準曲線に基づいて行いました。前処理されたサンプルはバルク溶液として調製され、一晩消化するために三連に分けられ、翌朝アッセイされました。前処理は、0.1% SDSと100 µg/ml Proteinase Kの存在下、55℃で一晩消化しました。
どちらの場合も、DNAのレベルはメソッドの検出限界以下であったと報告できます。これら2つのケースでは、サンプル成分は添加されたDNAの回収に影響せず、その結果、正確な定量が検証されました。
複数のスパイクレベルにおける許容可能な回収率に基づいてアッセイの定量精度が検証されれば、サンプルとその適切な緩衝液コントロールの複製物(複製物や三複製物など)を試験し、その差を計算することによって、ルーチンの定量を行うことができます。しかし、ルーチン検査においても、内部コントロールとして1~2種類の適切なスパイクレベルを含めることが推奨されます。高感度が要求される場合、または検出限界が問題となる場合は、標準曲線のローエンドにある複数のスパイクレベルでの複製物の試験が推奨されます。これにより、Y-切片とその結果生じる残留DNAレベル(サンプルと緩衝液コントロールライン間のY-切片の差)をより高い信頼性で決定することができます。
検出限界
閾値システムを用いたゼロキャリブレーター中の DNA の検出限界は 2 pg ± 1 pg2 です。バイオ医薬品サンプルを検査する場合、通常、サンプルの干渉を適切に除去するための前処理方法を採用する必要があります3 。前処理方法とサンプル成分はシステムの検出限界に影響します。検体の検出限界と必要な前処理は、標準曲線の下限(10 pg以下)の複数のスパイク・レベルで反復試験を行うことにより決定することができる。表 1 は、一晩のプロテアーゼ消化後、1.6 mg のプロテイン X 中の DNA を測定する場合の検出限界の決定方法の一例を示しています。
| サンプル | シグナル (µV/sec) |
標準偏差 偏差 |
CV (%) |
|---|---|---|---|
| 1.6 mg タンパク質 X + 0 pg | 35.5 | 2.3 | 6.5 |
| 1.6 mg タンパク質 X + 2 pg | 50.5 | 4.3 | 8.5 |
| 1.6 mg タンパク質 X + 4 pg | 61.9 | 5.9 | 9.5 |
| 1.6 mg タンパク質 X + 5 pg | 67.8 | 7.3 | 10.8 |
| 1.6 mg タンパク質 X+10 pg | 95.2 | 3.5 | 3.7 |
| 1.6 mg タンパク質 X + 20 pg | 168.0 | 9.7 | 5.8 |
表1:検出限界の決定例
イムノアッセイ開発では、検出限界の信頼水準について以下のような定義が一般的に用いられている:
99.9%信頼水準:陽性-2標準偏差。> 陰性 + 2 std.
95%信頼水準:陽性-1標準偏差。> 陰性+1標準偏差
これらの定義を使って、このサンプルについて以下の計算ができる:
2 pg - 2 std. dev. = 50.5 - 8.6 = 41.9 > 40.1 = 35.5 + 4.6 = 0 pg + 2 std. dev.
この結果に基づき、1.6mgのタンパク質Xは、一晩の消化を使用して、99.9%の信頼性で2pgの検出限界でアッセイできます。従って、このサンプルの残留DNAの定量を報告する場合、サンプルと適切な対照バッファーとの間に差が見られない場合、残留DNAのレベルは、1.6mgタンパク質あたり2pg DNA未満、または1.25pg/mgタンパク質未満として報告することができます。一般に、一晩の消化を必要とする前処理では、0.5mlの試料あたり2~5pgの範囲の検出限界が得られるが、一晩の消化に続いてDNA抽出を行うと、余分な試料の取り扱いが必要となるため、通常、0.5mlの試料あたり5~10pgの範囲の検出限界が得られます。
上記の分析では、スティック間のばらつき("スティックゲイン")を調整することなく、異なるスティックに散布された3連の "生 "シグナルを使用しました。これにより検出限界をより控えめに見積もることができます。通常、検出限界を決定する前に、まずスティックゲインで割って信号を調整するのが最善です。このゲイン調整は、スティックのゲイン値の範囲が大きい場合に重要になります。
まとめ
残留DNAは、複数のレベルで複製サンプルとコントロールをスパイクし、2つの直線(サンプルとコントロール)のy-切片の差を取ることによって得られたデータの線形回帰分析を使用して手動で定量することができます。この分析は、標準曲線の低い直線部分においてのみ有効です。分析に用いる値は、通常の標準曲線を用いた定量に基づくことができます。したがって、標準曲線および関連するオン・スティック・キャリブレーターの前処理は必要なく、一般に推奨されません。前処理法自体に関連するバックグラウンド以上のレベルを測定した場合は、DNase I 処理および/またはサンプルの希釈系列で確認する必要があります。
本アプリケーションノートでは、最終製品であるバイオ医薬品サンプル中の残留 DNA 濃度を測定するために使用される検査法の定量精度を検証するための一つの手法を示します。関連する前処理法を用いて、特定の試料の残留 DNA 量と検出限界の両方を測定する方法についての提案が示されています。これらの提案は、全てのユーザーが従わなければならない厳密なガイドラインと考えるべきではなく、閾値システムを効率的に適用するためのガイドラインとしてのみ捉えるべきです。
参考文献
1 Williams, D. F. 試験法バリデーションの概要。BioPharm Mfg 1:34-36, 51(1987).2 Kung V. T.、Panfili, P.R.、Sheldon, E. L.、King, R. S.、Nagainis, P. A.、Gomez, B.、Ross, D. A.、Briggs, J.、およびZuk, R. F. シリコンセンサーベースのシステムにおけるDNA結合タンパク質を用いた全DNAのピコグラム定量。Anal. Biochem. 187:220- 227 (1990).
3 Threshold Total DNA アプリケーションノート「高タンパク、低 DNA サンプルを Threshold Total DNA 分析に適応させる」、Molecular Devices Corporation (1989).
4 Threshold Total DNA アプリケーションノート「Threshold Total DNA アッセイで使用する DNA 標準試料の調製と適格性確認」、Molecular Devices Corporation (1990).
PDF版(英語)