Application Note マイクロ流体プラットフォームで
ニューロン・グリア3Dネットワークの
ハイスループット化合物評価
PDF版(英語)
著者一覧
Nienke R. Wevers *1,2、Remko van Vught *1、Karlijn J. Wilschut *1、Arnaud Nicolas *1,3、Chiwan Chiang *1、Henriette L. Lanz *1、Sebastiaan J. Trietsch *1、Jos Joore *1、Paul Vulto *1,3
in vitro脳モデル分野の大きな進歩により、現在の課題は、これらの技術を新しい薬剤候補の開発と評価に実装することです。ここでは、マイクロ流体プラットフォーム上で自発的に活動するニューロンと、それを支持するグリア細胞の3Dネットワークを培養する方法を示します。このプラットフォームはハイスループット特性を備え、標準的な実験機器との互換性があるため、化合物の効果を並列評価できます。
中枢神経系(CNS)疾患の有病率は増加していますが、多くの一般的な疾患において病態メカニズムの理解は依然として非常に限られており、その結果、効果的な治療法が不足しています。長い間、生理学的に関連性のあるヒト脳のin vitroモデルの欠如が、新薬候補の開発における主要なボトルネックでした。しかし、iPS細胞(iPSC)技術とそれに伴う分化プロトコールの利用可能性により、この分野は大きく進展しました *1,2。現在のプロトコールでは、ヒト脳のほぼすべての細胞サブタイプへの分化が可能であり、さまざまなニューロンのサブタイプ *3-7、グリア細胞 *8-10、内皮細胞 *11が含まれます。
同時に、発生生物学の分野では、幹細胞、分化細胞、細胞外マトリックス(ECM)の3D in vivo構造をin vitroで成長させたオルガノイドで再現することに大きな進歩がありました *12-14。最近では、ECMにカプセル化されたiPSC由来の神経幹細胞が、大脳オルガノイドに発達し、新生児脳のさまざまな特徴を模倣することが示されています *15。この技術は、脳の発達や神経疾患の病因研究に急速に導入されました *16。
脳の生理機能を模倣するこれらの大きな進歩により、薬剤候補や化合物評価を日常的に行うために、これらの技術をどのように実装するかという課題が生じています。化合物ライブラリーの有効性と毒性の影響を評価できるシステムが必要です。このようなプラットフォームの重要な前提条件には、自動ハイコンテントイメージング装置との互換性と比較的迅速な読み出しが含まれます。さらに、モデルは、各データポイントに必要な細胞材料の量を最小限に抑えながら、ヒト脳の複雑性を生理学的に関連性のある応答を得るために必要な範囲で再現する必要があります。
ここでは、OrganoPlate®と呼ばれるマイクロ流体プラットフォームで、ECMに埋め込まれたニューロン・グリアの3Dネットワークを培養する方法を示します。OrganoPlate®は、96個のティッシュチップを備えたマイクロタイタープレートフォーマットで、3D細胞培養、共培養、非侵襲的な培地交換に使用できます *17。ヒトiPSC由来の神経幹細胞、またはさまざまなソースからのiPSC由来成熟ニューロンとアストロサイトをMatrigel®と混合し、これらのマイクロ流体チップに播種します。細胞はチップ内で3Dネットワークを形成し、免疫蛍光染色を用いて特徴付けられます。さまざまな化合物の潜在的な神経毒性効果は、ネットワーク内のニューロンの電気生理学的活動、神経突起伸長の程度、および化合物処理に対する細胞の生存率を評価することで研究されました。
結果
プラットフォーム 図1はOrganoPlate®プラットフォームを示しています。このプラットフォームは384ウェルマイクロタイタープレートフォーマットに基づき、光学アクセスのためにカバーガラス厚(175 μm)のガラスを採用しています。プレートには96個のマイクロ流体ティッシュチップが含まれ、それぞれが小型化されたティッシュモデルを構築するために使用できます *17-19。各チップは4つの隣接するウェルを接続します:1つのウェルは細胞/ECM混合物の投与に使用され、2つのウェルは培養培地の供給に、4つ目のウェルはイメージングに使用されます(図1a)。細胞/ECM混合物は、フェーズガイドと呼ばれる毛細管圧バリアを用いてティッシュチップのゲルチャンネルにパターン化されます *20。ゲル化後、隣接するチャンネルに培養培地が充填され、栄養素、ガス、老廃物の交換が妨げられずに行われます(図1b)。培地のインレットとアウトレットは、培地の交換や化合物および染色試薬の細胞への投与に使用できます。
ヒト脳のin vitroモデルを実現するために、iPSC由来のニューロンとグリア細胞の3D ECM埋め込みネットワーク形成のための培養条件を最適化しました。さまざまな分化段階の異なるタイプのニューロン細胞を使用し、プラットフォームのニューロン培養との互換性を評価し、その可能な応用を探りました。神経幹細胞や初期分化細胞は、発達過程、疾患メカニズム、患者特異的プロセスの研究に興味深い長期3Dニューロン分化のオプションを評価するために使用されました。OrganoPlate®に播種時にすでに終末分化状態にある他の細胞は、化合物スクリーニングなどの用途に便利な短期培養のオプションを評価するために使用されました。本研究で使用した異なる細胞タイプの概要は表1に示されています
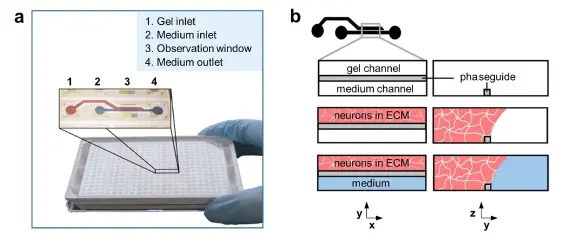
図1. OrganoPlate®へのニューロンとグリアの播種 (a) OrganoPlate®の写真。OrganoPlate®は384ウェルプレートを基盤とし、合計96個のマイクロ流体ティッシュチップで構成されています。インレイは単一チップのイメージ図を示しており、各チップは4つのウェルで構成されています:ゲルインレット(1)、培地インレット(2)、観察窓(3)、培地アウトレット(4)。これらはマイクロ流体チャンネルで接続されています。(b) 3Dニューロン・グリアネットワーク培養の実験概要:細胞は細胞外マトリックス溶液(ピンク)と混合され、ゲルインレットウェル(1)にピペッティングしてゲルチャンネルに播種されます。フェーズガイドは毛細管圧バリアであり、ゲルをゲルチャンネルに制限します。ゲル化後、細胞培養培地(青)を培地インレットとアウトレットウェルに添加し、培地チャンネルを満たし、膜を介さない栄養素、代謝物、ガスの直接交換を可能にします。
| 名前 | サプライヤー | 特徴 |
|---|---|---|
| Axol neural stem cells | アクソルバイオサイエンス | ハンガントン病患者由来のヒトiPSC由来神経幹細胞。約6週間の分化期間を要します。 |
| HIP ™ neurons | アムスビオ | 健常ドナー由来のヒトiPSC由来初期分化ニューロンおよびアストロサイト。約5週間の分化期間を要します。 |
|
Dopa.4U ™ neurons |
アクシオジェネシス | 健常ドナー由来のヒトiPSC由来終末分化ニューロン。これらのニューロンの亜集団はドーパミン作動性です。 |
| iCell® neurons | セルラー・ダイナミクス・インターナショナル | 健常ドナー由来のヒトiPSC由来終末分化ニューロン。このニューロンは主にGABA作動性およびグルタミン酸作動性です。 |
| iCell® astrocytes | セルラー・ダイナミクス・インターナショナル | 健常ドナー由来のヒトiPSC由来アストロサイト。 |
表1. 本研究で使用したiPSC由来細胞の概要と特性
3Dネットワークの迅速な形成 神経幹細胞、初期分化細胞、またはアストロサイトを含まない成熟ニューロンをMatrigel®と混合し、OrganoPlate®のゲルチャンネルに播種しました。細胞はECM内で迅速にネットワークを形成し、ニューロン形態を示します。図2aは、終末分化したドーパミン作動性Dopa.4U™ニューロンのネットワーク形成の経時変化を示しています。培養は播種後の異なるタイムポイントで固定され、ニューロンマーカーβ3-チューブリンで染色されました。神経突起伸長はタイムポイントゼロに対して定量化され、迅速なネットワーク形成を示しました(図2b)。ネットワークの三次元構造は共焦点イメージングで確認されました(図2cおよび補足動画1)。終末分化ニューロンとグリアの共培養でも同様のネットワーク形成が観察されました(iCell®ニューロンとアストロサイト、補足動画2)。
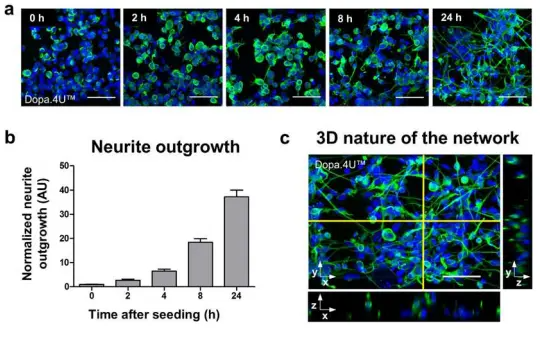
図2. 3Dネットワークの形成 (a) ECM溶液と混合したDopa.4U™ニューロン(Axiogenesis)をティッシュチップに播種し、ネットワーク形成を許可しました。細胞は播種後の異なるタイムポイント(0、2、4、8、24時間)で固定され、β3-チューブリンおよびDNA(DraQ5)染色によりネットワークと核が可視化されました。スケールバー:50 μm。(b) 神経突起伸長の経時的定量化(t = 0 hに正規化、平均 ± SEM、SEMは代表的な単一培養チップ内の200–400細胞に基づく)。 (c) OrganoPlate®内のDopa.4U™ニューロン培養(1日目)の最大投影像。β3-チューブリンおよびDraQ5で染色。右と下の画像は選択平面の直交ビューを示します。
OrganoPlate®はさまざまなニューロンおよびグリアサブタイプの培養をサポート ヒト脳は、さまざまなニューロンやグリア細胞を含む無数の細胞タイプで構成されています。本研究では、3DシステムがiPSC由来のグルタミン酸作動性、GABA作動性、およびドーパミン作動性ニューロンの培養をサポートするかを調べました。さらに、終末分化したiPSC由来アストロサイトを用いてニューロン・グリア共培養を確立し、ヒト脳の複雑性をさらに模倣しました。図3aは、培養2週間後のiCell®ニューロンとアストロサイトの混合集団を示しています。2種類の細胞は、それぞれβ3-チューブリン(ニューロン)とグリア線維性酸性タンパク質(GFAP;アストロサイト)を明確に発現しています。図3b–dは、OrganoPlate®で6週間分化したAxol iPSC由来神経幹細胞から得られたニューロン・グリアネットワークを示しています。ニューロンマーカーβ3-チューブリンとアストロサイトマーカーvimentinおよびs100βの発現により、ニューロンとアストロサイトの混合集団が得られたことが示されています(図3b,c)。さらに、ニューロン集団にはグルタミン酸作動性(vGLUT)およびGABA作動性(vGAT)ニューロンが含まれており(図3d)、興奮性細胞と抑制性細胞の両方が存在することを示しています。図3eは、終末分化したDopa.4U™ニューロン(β3-チューブリン)の免疫蛍光染色を示しており、その一部はドーパミン合成の律速酵素であるチロシンヒドロキシラーゼ(TH)を発現しています。
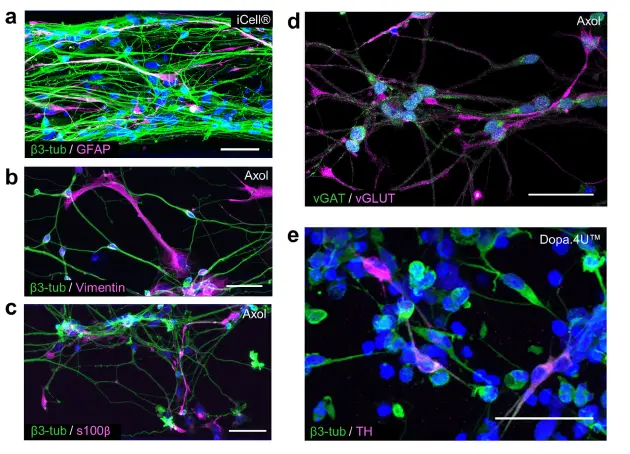
図3. OrganoPlate®はさまざまな脳細胞タイプの成長と分化をサポート (a–c) 免疫蛍光画像の最大投影は、ニューロンとアストロサイトの混合集団を示します。(a) 成熟したiCell®ニューロン(β3-チューブリン)とアストロサイト(GFAP)の共培養(14日目)。(b,c) Axol Huntington神経幹細胞は、OrganoPlate®で6週間後にニューロン(β3-チューブリン)とアストロサイト(vimentin、s100β)に分化。スケールバー:50 μm。(d,e) 免疫蛍光画像の最大投影は、異なるニューロンサブタイプの存在を示します:(d) Axol神経幹細胞は、6週間の分化後にグルタミン酸作動性(vGLUT)およびGABA作動性(vGAT)ニューロンを示す。 (e) Dopa.4U™ニューロン(5日目)、β3-チューブリンおよびチロシンヒドロキシラーゼ(TH)で染色。スケールバー:50 μm。
自発的なニューロン発火 自発的な電気生理学的活動は、カルシウムイメージングを用いて各細胞ソースで研究され、単一細胞およびネットワーク全体のニューロン活動の調査を可能にしました。カルシウム感受性蛍光色素Fluo-4 AMを細胞にロードし、高速イメージングを用いて細胞内カルシウム変動を経時的にモニタリングしました(図4a–c)。終末分化ニューロン(Dopa.4U™およびiCell®)と、3Dで分化したニューロン細胞(Axol、HIP™)の両方で、自発的な電気生理学的活動が確認されました。各細胞ソースについて、代表的な活動ニューロンを選択し、蛍光シグナルを時間経過でプロットしました(図4d–g)。
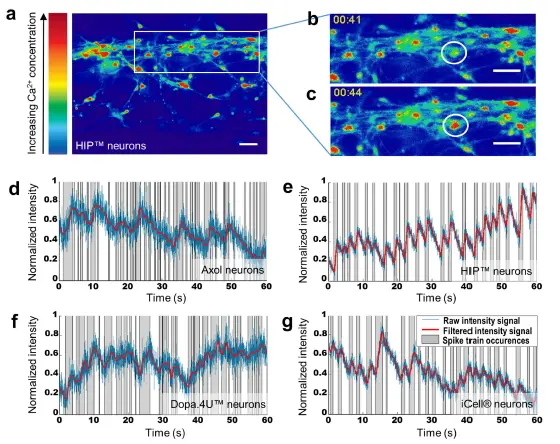
図4. カルシウムイメージングは、すべての細胞ソースで自発的なニューロン活動を示す (a) OrganoPlate®で5週間分化したHIP™ニューロンのカルシウムイメージング中に取得された画像の例。色の暖かさは細胞内カルシウム濃度に対応します。(b,c) 1つの細胞における細胞内カルシウム変動の例(t = 41秒およびt = 44秒)。スケールバー:50 μm。(d–g) 各細胞ソースから活動細胞を選択し、その細胞内蛍光を時間経過でプロットしました。各細胞の蛍光強度は、その細胞の最小および最大蛍光信号に基づき0から1にスケーリングされ、ローパスフィルターで処理されています。グラフは漂白補正後の生データ(青)、フィルタ処理後の信号(赤)、および局所的な最小値と最大値の間の活動領域(灰色)を示し、異なるスパイク列を表します。
3Dニューロン・グリアネットワークにおける化合物効果の評価
電気生理学 - 3D培養のハイスループット化合物評価への適用性を評価するため、電気生理学、神経突起伸長、および細胞生存率に対する化合物の影響を調べました。カルシウムイメージングを用いて、化合物添加前後のニューロンの電気生理学的活動を検出しました(図5a、補足動画3–6)。iCell®ニューロンをOrganoPlate®に播種し、7日間培養後、GABA(100 μM)またはテトロドトキシン(TTX、1 μM)に曝露しました。GABAとTTXはいずれもiCell®ニューロンの発火を阻害しましたが、コントロールチップでは発火パターンに変化は見られませんでした(図5b–d)。GABAは成熟脳における主要な抑制性神経伝達物質ですが、神経発達中は興奮性に作用します *21,22。OrganoPlate®内の3D iCell®ニューロン培養におけるGABAの抑制効果は、このネットワークが成熟ニューロン集団で構成されていることを改めて示しています。TTXはフグに存在する強力な神経毒であり、電位依存性ナトリウムチャネルを遮断し、活動電位の発火を阻害することが知られています²³。
細胞生存率 - 次に、ニューロンとアストロサイトの共培養において、3種類の神経毒性化合物のさまざまな濃度への曝露後の細胞生存率を評価しました。iCell®ニューロンとアストロサイトを同数でMatrigel®と混合し、OrganoPlate®で6日間培養後、化合物に24時間曝露しました。RealTime-Glo™ Cell Viabilityアッセイを用いて、細胞の還元能、すなわち生存率を測定しました。試験した3種類の既知の神経毒性物質(メチル水銀 *24、エンドスルファン *25、2,5-ヘキサンジオン *26)は、濃度依存的に細胞生存率を低下させました(図5e)。
神経突起伸長 - さらに、化合物毒性を検出するために神経突起伸長を評価しました。Dopa.4U™ニューロンをOrganoPlate®に播種し、24時間ネットワーク形成を許可しました。その後、酸化ストレスを誘発することが知られている神経毒性化合物メチル水銀 *24の濃度範囲に曝露しました。24時間曝露後、培養を固定し、β3-チューブリン染色によりニューロンネットワークを可視化しました。メチル水銀濃度の増加に伴い、平均神経突起長の有意な減少が観察されました(図5f,g)。
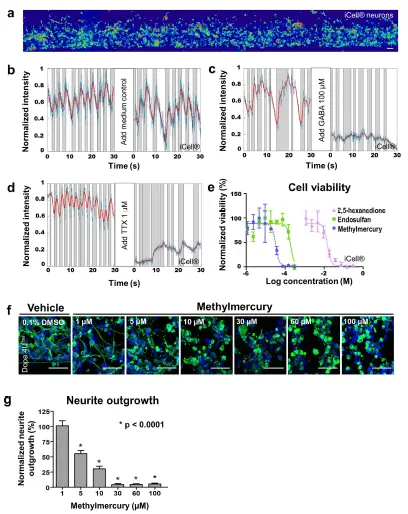
図5. 電気生理学、神経突起伸長、および細胞生存率に対する化合物効果の検出 (a) カルシウムイメージング記録は、化合物添加前後の電気生理学的活動を評価するため、iCell®ニューロン(播種後7日目)のネットワーク全体を捉えています。色の暖かさは細胞内カルシウム濃度に対応します。スケールバー:50 μm。 (b–d) グラフは漂白補正後の生信号(青)、フィルタ処理後の信号(赤)、および局所的な最小値と最大値の間の活動領域(灰色)を示し、異なるスパイク列を表します。GABA(100 μM)またはTTX(1 μM)の添加は、iCell®ニューロン(播種後7日目)の発火を阻害します。 (e) iCell®ニューロンとiCell®アストロサイトを同数でECM溶液に混合し、OrganoPlate®に播種しました。6日間培養後、細胞はメチル水銀、エンドスルファン、または2,5-ヘキサンジオンのさまざまな濃度に24時間曝露され、その後RealTime-Glo™アッセイ(Promega)を用いて細胞生存率を評価しました。細胞生存率は、ビークルコントロール(メチル水銀およびエンドスルファンには0.3% DMSO、2,5-ヘキサンジオンには培地)と比較してプロットされています(平均 ± SD、n = 3)。 (f) Dopa.4U™ニューロンをECM溶液に混合し、OrganoPlate®に播種して24時間ネットワーク形成を許可した後、メチル水銀のさまざまな濃度に24時間曝露しました。細胞は固定され、β3-チューブリンおよびDraQ5で染色し、神経突起と核を可視化し、共焦点顕微鏡で撮影しました。最大投影画像を示します。スケールバー:50 μm。 (g) メチル水銀に曝露されたニューロンの神経突起伸長の定量化は、濃度依存的な減少を示しました(平均 ± SEM、SEMは代表的な単一培養チップ内の200–400細胞に基づく、Kruskal-Wallis検定(非ガウスデータセット)およびDunnの事後検定による多重比較、*p < 0.0001)。
ディスカッション
本論文では、3Dニューロン・グリアネットワークの並列培養と試験のための方法を紹介しました。ネットワークはマイクロ流体チャンネル内で成長し、さまざまな商業ソースからのiPSC由来ニューロンとアストロサイトで構成されています。これらの培養の3D構造は、現代のin vitroモデリングに不可欠な前提条件です。分化、遺伝子発現、細胞間相互作用、細胞-マトリックス相互作用など、多くの重要な細胞プロセスや特性は、in vivoと比較して2D平面設定では異なるため、2D細胞培養から得られた結果を生体に外挿することは困難です *27-30。3D培養システムで成長したニューロンは、2D単層と比較して、より長い神経突起伸長、細胞生存率の改善、および異なる分化パターンを示すことが観察されています *31,*32。
一方、現在の3D培養システムにはいくつかの欠点があります。現在の3D細胞培養および脳オルガノイドシステムは、拡散輸送の制限、播種条件の大きなばらつき、細胞播種、培地交換、染色、イメージングの複雑な手順に悩まされています *30-33。本研究で導入したプラットフォームは、3D培養に関連する取り扱いと再現性の問題を大幅に解決します。96個のチップを同時に播種することでウェル間のばらつきを減らし、ポンプを必要としないユーザーフレンドリーな設計です。小容量、マイクロ流体ゲル層化、および多孔性細胞外マトリックスの組み合わせにより、拡散輸送の制限を防ぎます。このシステムでは流れは積極的に誘導されませんが、マルチリザーバーマイクロ流体チャンネルシステムに固有の最小限の流れが存在し、栄養供給と老廃物除去を拡散だけでなく細胞外マトリックスを介した間質流によっても可能にします。さらに、培地リザーバーとマイクロ流体チャンネル内の実際の培養容量との幾何学的分離により、培地交換や化合物・染色試薬の添加は完全に非侵襲的であり、マイクロ流体空間での補充はピペッティングではなくリザーバー間の受動的なレベリングによって行われます。他のマイクロ流体培養システムと比較して、本プラットフォームには人工膜が完全に存在せず、ECM間質液と培地チャンネルの交換が妨げられません *17-20。明らかに、このプラットフォームのハイスループット特性と標準的な実験機器との互換性、ならびに各ティッシュチップに必要な細胞材料の量が限られていることにより、さまざまな濃度で化合物を複数のレプリケートで評価できます。さらに、このプラットフォームは自動化環境に容易に適応でき、より多くの化合物や条件のスクリーニングを可能にします。
OrganoPlate®内のニューロン・グリア培養の3D構造は、すべての方向への細胞成長とネットワーク形成を可能にし、細胞-マトリックス相互作用を実現し、従来の2D培養と比較して複雑性を高めます。ただし、本研究で提示したモデルは、ヒト脳に存在するすべての細胞タイプを網羅しているわけではありません。目的に応じて複雑性を高めるために、追加の細胞タイプをモデルに組み込むことができます。オリゴデンドロサイトやミクログリアなどの他のグリア細胞を追加して、中枢神経系内の髄鞘形成や免疫プロセスを研究できます。さらに、さまざまな疾患の患者由来iPSC細胞をOrganoPlate®で分化させ、発達異常や疾患プロセスを研究することも可能です。
もう1つの選択肢は、ニューロン・グリアネットワークに隣接する培地チャンネルで脳微小血管ペリサイトと内皮細胞の血管を成長させ、血液脳関門(BBB)を模倣することです *34。BBBは、中枢神経系の恒常性環境を確保する高度に特殊化されたバリアです。親水性化合物の受動拡散を制限することで、BBBはほとんどの薬剤が脳に到達するのを妨げます。したがって、アストロサイトとニューロンに加えてBBBを含むモデルは、薬剤候補評価をさらに改善します。これにより、脳内での化合物効果の評価だけでなく、化合物が脳に侵入できるかどうか、BBBの完全性に影響を与えるかどうかも評価できます。
最後に、パーソナライズド医療はCNS疾患治療の未来を代表します *35。ほとんどの疾患は患者ごとに異なる原因によって生じるため、これらの疾患を効果的に標的とするには、オーダーメイド治療が必要になる可能性があります。iPSC技術を使用することで、本手法は特定患者の96個のニューロン・グリアネットワークを培養し、さまざまな化合物を試験して最適な治療オプションを評価でき、治療効果を高め、副作用を減らすことができます。
以上のことから、私達は、マイクロ流体プラットフォームにおいて、ニューロンおよびグリアの3Dネットワーク上で医薬品化合物のハイスループット評価を可能にする新規手法を開発しました。様々な種類の細胞を用いて、このプラットフォームが、神経幹細胞や前駆細胞の長期分化や、定義された比率で混合可能な終末分化細胞の短期培養に適合することを示しました。ネットワークはロバスト性と時間効率の高い方法で形成され、細胞播種後7日以内にアッセイを行うことができます。この3D ECM包埋ネットワークは、カルシウムイメージング、神経突起伸長、細胞生存率など、幅広いリアルタイムおよびエンドポイントアッセイとの適合性を示しました。このプラットフォームの使いやすさと標準的な実験装置との互換性により、化合物のスクリーニングや評価を目的とした3D中枢神経系モデルの可能性が最大限に引き出されました。最後に、患者由来のiPSCを用いることで、この方法は、中枢神経系の一般的な疾患の治療を改善するための個別化医療の選択に使用することができます。
方法
材料
以下の試薬はThermo Fisher Scientific(米国マサチューセッツ州ウォルサム)から購入しました:B-27®サプリメント50x、Fluo-4 AM細胞透過性色素、ホルムアルデヒド37%、GlutaMAX™サプリメント、ヒト組換え上皮成長因子(EGF)、MEM非必須アミノ酸溶液100x、N-2サプリメント100x、リン酸緩衝生理食塩水1x(PBS)。 以下の試薬はSigma-Aldrich(米国ミズーリ州セントルイス)から購入しました:2,5-ヘキサンジオン、γ-アミノ酪酸(GABA)、ウシ血清アルブミン(BSA)、ジメチルスルホキシド(DMSO)、Dulbecco’s Modified Eagle Medium(DMEM)、エンドスルファン、メチル水銀、ペニシリン-ストレプトマイシン溶液100x、Triton™ X-100、TWEEN® 20。 Matrigel®はCorning(米国ニューヨーク州コーニング)から購入しました。胎児ウシ血清(FCS)はATCC(米国バージニア州マナサス)から購入しました。ヒト組換え線維芽細胞成長因子(bFGF)はPeprotech(米国ニュージャージー州ロッキーヒル)から購入しました。テトロドトキシン(TTX)はTocris Bioscience(英国ブリストル)から購入しました。
細胞培養
本研究では、4種類の市販ニューロンおよびグリア細胞ソースを使用しました。神経幹細胞(ハンチントン病患者由来)はAxol Bioscience(英国ケンブリッジ)より提供され、HIP™ニューロンはAMSBIO(英国アビンドン)から購入しました。Dopa.4U™ニューロンはAxiogenesis(ドイツ・ケルン)、iCell®ニューロンおよびアストロサイトはCellular Dynamics International(米国ウィスコンシン州マディソン)から購入しました。 すべてのニューロン細胞タイプは、供給元が提供する培地に100 U/mLペニシリンおよび100 μg/mLストレプトマイシンを添加して培養しました。iCell®アストロサイトは、Matrigel®コートフラスコ(DMEM中80 μg/mLのMatrigel®を37°C、5% CO₂で15分間インキュベート)で培養し、DMEMに15% FCS、2 mM GlutaMAX™、1%非必須アミノ酸溶液、100 U/mLペニシリン、100 μg/mLストレプトマイシン、10 μg/mL bFGF、10 μg/mL EGFを添加して、OrganoPlate®に播種する前に4パッセージ行いました。すべての神経細胞は、解凍および遠心後すぐにOrganoPlate®に播種しました。
OrganoPlate®でのニューロンおよびニューロン-グリア培養の播種と培養 細胞はメーカーの指示に従って解凍し、遠心しました。遠心後、上清を除去し、細胞を7 mg/mLのMatrigel®(9.3 mg/mLのMatrigel®ストックを冷DMEMで希釈)に再懸濁し、最終濃度25,000 cells/μLとしました。リピーティングピペット(Sartorius eLINE電子ピペット)を使用し、各OrganoPlate®ティッシュチップのゲルチャンネルに細胞懸濁液0.5–1 μLを播種しました(MIMETAS、オランダ)。その後、OrganoPlate®をインキュベーター(37°C、5% CO₂)に20分間置き、Matrigel®のゲル化を許可しました。次に、培地インレットに50 μLの培地(供給元提供の培地に100 U/mLペニシリンおよび100 μg/mLストレプトマイシンを添加)を加え、続いて培地アウトレットに50 μLの培地を加え、OrganoPlate®をインキュベーターに戻しました(図1b参照)。培地交換は週2回、培地インレットおよびアウトレットウェルから半量を除去し、新しい培地25 μLを追加することで行いました。培養の進行と一貫性を評価するため、OrganoPlate®の96チップすべてから位相差画像を週2~3回取得しました(ImageXpress Micro XLSハイコンテントイメージングシステム、モレキュラーデバイス、米国カリフォルニア州サニーベール)。
免疫細胞化学
各ステップは1チップに使用する手順と容量を記載しています。各ステップで、特記がない限り、50 μLの溶液をチップの培地インレットおよびアウトレットに添加します。ゲルインレットはすべての手順で空のままにします。すべてのステップは室温(RT)で実施します。各洗浄ステップは5分間です。培地を除去し、PBSで細胞を5分間洗浄しました。PBSを除去し、3.7%(v/v)ホルムアルデヒドPBS溶液で15分間固定しました。固定液を除去し、PBSで3回、4% FCS(v/v)PBS溶液で1回洗浄しました。次に、0.3%(v/v)Triton X-100 PBS溶液で10分間透過化し、4% FCS PBS溶液で1回洗浄しました。細胞を2%(v/v)FCS、2% BSA(w/v)、0.1%(v/v)TWEEN® 20 PBS溶液で45分間ブロッキングし、一次抗体溶液(チップあたり40 μL;培地インレットに10 μL、アウトレットに30 μL)で室温1時間または4°Cで一晩インキュベートしました。以下の一次抗体をブロッキング溶液で希釈し、免疫細胞化学に使用しました:β3-チューブリン(1:200, Abcam, AB78078)、β3-チューブリン(1:200, Abcam, AB18207)、GFAP(1:200, Millipore, AB5804)、Vimentin(1:20, Abcam, AB8978)、S100β(1:100, Abcam, AB52642)、vGLUT(1:1000, Abcam, AB72310)、vGAT(1:750, Synaptic systems, 131011)、チロシンヒドロキシラーゼ(1:2000, Santa Cruz, H-196)。ウェルを4% FCS PBS溶液で3回洗浄し、続いて二次抗体溶液40 μL(培地インレットに10 μL、アウトレットに30 μL)を添加しました。以下の二次抗体を使用しました:Alexa Fluor® goat-anti-mouse 488、Alexa Fluor® goat-anti-rabbit 488、Alexa Fluor® goat-anti-mouse 555、Alexa Fluor® goat-anti-rabbit 555(Thermo Fisher、ブロッキング溶液で1:250に希釈)。暗所で30分間インキュベート後、ウェルを4% FCS PBS溶液で3回、PBSで1回洗浄しました。DraQ5(Abcam、PBSで1:1000希釈、培地インレットに10 μL、アウトレットに30 μL)を添加し、核を染色し、20分間インキュベートしました。DraQ5溶液を除去し、PBSに置換しました。染色細胞の高品質Zスタック画像は、Leica TCS SP5共焦点顕微鏡(Leica、ドイツ・ヴェッツラー)で取得しました。
カルシウムイメージング
各ステップは1チップに使用する手順と容量を記載しています。各ステップで、特記がない限り、50 μLの溶液をチップの培地インレットおよびアウトレットに添加します。ゲルインレットはすべての手順で空のままにします。Fluo-4 AM色素を用いてカルシウムイメージングにより自発的なニューロン活動を記録しました。選択したウェルの培地を、培地中の5 μM Fluo-4 AM溶液(最終DMSO濃度0.1%)に置換し、室温で30分間インキュベートしました。次に、Fluo-4溶液を色素を含まないコンディショニング培地に置換し、室温で15分間インキュベートしました。蛍光信号の変化を検出するため、高速イメージングをLeica AF6000顕微鏡で実施しました(20倍、50 Hz、ビニング2×2)。各細胞ソースについて、複数の活動細胞を解析し、代表的な記録を選択して蛍光信号を時間経過でプロットしました。カルシウム記録はFijiの漂白補正プラグインを使用してフォトブリーチングを補正しました *36。選択した各細胞の信号は0から1にスケーリングされ、MATLAB®(2013bおよびSignal Processing Toolbox、The MathWorks Inc、米国マサチューセッツ州ナティック)でローパスフィルター(Butterworth、0.5 Hz、3次)を通しました。結果は漂白補正後の生信号(青)、フィルタ処理後の信号(赤)、および局所的な最小値と最大値の間の活動領域(灰色)を示し、異なるスパイク列を表します(図4d–g)。
カルシウムイメージング化合物アッセイ
iCell®ニューロンは、前述の方法でFluo-4 AMをロードしました。イメージング直前に、化合物添加時の希釈を最小限に抑えるため、培地インレットおよびアウトレットウェルから培地を除去しました。マイクロ流体チップ内に残るサブマイクロリットル量の培地は、細胞への影響を避けるため除去しませんでした。自発的な活動は、ImageXpress Micro XLS-C ハイコンテントイメージングシステム(モレキュラーデバイス、ワイドフィールドモード、20倍、20 Hz)を用いて30秒間記録しました。画像取得を一時停止し、100 μM GABAまたは1 μM TTXを含む培地50 μLを培地インレットおよびアウトレットウェルに慎重にピペッティングしました。画像取得は30秒後に再開し、化合物効果を記録しました。化合物添加前後の記録は、Fijiの漂白補正プラグイン *36を用いて個別に補正しました。このプラグインは、各記録のすべてのフレームの平均強度を最初のフレームに補正します。2回目の記録開始時(化合物条件後30秒)の高度な漂白を補正するため、2回目の記録の平均強度を両記録の平均強度比で乗算しました。漂白補正後の記録を連結し、関心領域を手動で選択して複数細胞の蛍光信号を時間経過でプロットしました。代表的な細胞の信号は、前述の方法で処理・プロットしました(図5b–d)。
神経突起伸長
Dopa.4U™ニューロンは、前述の方法でOrganoPlate®に播種し、播種後の異なるタイムポイント(0、2、4、8、24時間)で固定しました(免疫細胞化学セクション参照)。ニューロンはβ3-チューブリンおよびDraQ5で染色し、神経突起と核を可視化し、Leica TCS SP5共焦点顕微鏡で撮影しました(図2a)。核のみ、または核+β3-チューブリン染色の最大投影画像は、モレキュラーデバイス MetaXpressソフトウェア(MetaXpress 6.1)の神経突起伸長アプリケーションで解析しました。細胞または神経突起検出のエラーは手動で修正し、偽陽性値はピクセル長0に置換しました。グラフはGraphPad Prism 6(GraphPad Software、米国カリフォルニア州サンディエゴ)でプロットしました(図2b)。
化合物効果による神経突起伸長
Dopa.4U™ニューロンは、前述の方法でOrganoPlate®に播種し、24時間ネットワーク形成を許可しました。次に、細胞を完全なDopa.4U™培地中でメチル水銀またはビークルコントロール(0.3% DMSO、v/v)に24時間曝露しました。細胞はその後染色・撮影され、神経突起伸長は前述の方法で解析しました(図5f)。各条件の神経突起伸長はビークルコントロール(0.3% DMSO)に正規化しました。グラフはGraphPad Prism 6でプロットし、統計解析を実施しました。神経突起伸長データセット(非ガウス分布)は、Kruskal-Wallis検定およびDunnの事後検定による多重比較で解析しました(図5g)。
化合物効果による細胞生存率
iCell®ニューロンとアストロサイトを1:1の比率でOrganoPlate®に播種し、6日間培養後、神経毒性化合物の濃度範囲に24時間曝露しました。メチル水銀およびエンドスルファンのビークルコントロールは0.3% v/v DMSO、2,5-ヘキサンジオンのビークルコントロールは培地でした。24時間後、細胞生存率はRealTime-Glo™ MTアッセイ(Promega、米国ウィスコンシン州マディソン)を用いてメーカーの指示に従って評価しました。発光信号はFluoroskan Ascent™マイクロプレート蛍光計(Thermo Scientific)を用いて5分ごとに測定しました。各チップについて、培地インレット、観察窓、培地アウトレットを測定し、平均値を算出しました。各条件に3チップを使用し、正規化された平均値と標準偏差をGraphPad Prism 6でプロットしました(図5e)。
参考文献
- Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–76 (2006).
- Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–72 (2007).
- Hu, B.-Y. & Zhang, S.-C. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat. Protoc. 4, 1295–304 (2009).
- Swistowski, A. et al. Efficient generation of functional dopaminergic neurons from human induced pluripotent stem cells under defined conditions. Stem Cells 28, 1893–904 (2010).
- Shi, Y., Kirwan, P. & Livesey, F. J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–46 (2012).
- Yu, D. X. et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem cell reports 2, 295–310 (2014).
- Erceg, S., Lukovic, D., Moreno-Manzano, V., Stojkovic, M. & Bhattacharya, S. S. Derivation of cerebellar neurons from human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. Chapter 1, Unit 1H.5 (2012).
- Krencik, R. & Zhang, S.-C. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat. Protoc. 6, 1710–7 (2011).
- Wang, S. et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12, 252–64 (2013).
- Ohgidani, M. et al. Direct induction of ramified microglia-like cells from human monocytes: dynamic microglial dysfunction in Nasu-Hakola disease. Sci. Rep. 4, 4957 (2014).
- Lippmann, E. S. et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 30, 783–91 (2012).
- Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–5 (2009).
- Huch, M. et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015).
- Takasato, M. et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–8 (2015).
- Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–9 (2013).
- Garcez, P. P. et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–8 (2016).
- Trietsch, S. J., Israëls, G. D., Joore, J., Hankemeier, T. & Vulto, P. Microfluidic titer plate for stratified 3D cell culture. Lab Chip 13, 3548–54 (2013).
- Moreno, E. L. et al. Differentiation of neuroepithelial stem cells into functional dopaminergic neurons in 3D microfluidic cell culture. Lab Chip 15, 2419–28 (2015).
- Jang, M., Neuzil, P., Volk, T., Manz, A. & Kleber, A. On-chip three-dimensional cell culture in phaseguides improves hepatocyte functions in vitro. Biomicrofluidics 9, 34113 (2015).
- Vulto, P. et al. Phaseguides: a paradigm shift in microfluidic priming and emptying. Lab Chip 11, 1596–602 (2011).
- Owens, D. F., Boyce, L. H., Davis, M. B. & Kriegstein, A. R. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 16, 6414–23 (1996).
- Leinekugel, X. et al. GABA is the principal fast-acting excitatory transmitter in the neonatal brain. Adv. Neurol. 79, 189–201 (1999).
- Kiernan, M. C., Isbister, G. K., Lin, C. S.-Y., Burke, D. & Bostock, H. Acute tetrodotoxin-induced neurotoxicity after ingestion of puffer fish. Ann. Neurol. 57, 339–48 (2005).
- Yee, S. & Choi, B. H. Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology 17, 17–26 (1996).
- Gant, D. B., Eldefrawi, M. E. & Eldefrawi, A. T. Cyclodiene insecticides inhibit GABAA receptor-regulated chloride transport. Toxicol. Appl. Pharmacol. 88, 313–21 (1987).
- vDeCaprio, A. P., Briggs, R. G., Jackowski, S. J. & Kim, J. C. Comparative neurotoxicity and pyrrole-forming potential of 2,5-hexanedione and perdeuterio-2,5-hexanedione in the rat. Toxicol. Appl. Pharmacol. 92, 75–85 (1988).
- Hoffman, R. M. To do tissue culture in two or three dimensions? that is the question. Stem Cells 11, 105–111 (1993).
- Cukierman, E., Pankov, R., Stevens, D. R. & Yamada, K. M. Taking cell-matrix adhesions to the third dimension. Science 294, 1708–12 (2001).
- Pampaloni, F., Reynaud, E. G. & Stelzer, E. H. K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 8, 839–45 (2007).
- Ravi, M., Paramesh, V., Kaviya, S. R., Anuradha, E. & Solomon, F. D. P. 3D cell culture systems: advantages and applications. J. Cell. Physiol. 230, 16–26 (2015).
- Peretz, H., Talpalar, A. E., Vago, R. & Baranes, D. Superior Survival and Durability of Neurons and Astrocytes on 3-Dimensional Aragonite Biomatrices. Tissue Eng. 13, 461–472 (2007).
- Zare-Mehrjardi, N. et al. Differentiation of embryonic stem cells into neural cells on 3D poly (D, L-lactic acid) scaffolds versus 2D cultures. Int. J. Artif. Organs 34, 1012–23 (2011).
- Kelava, I. & Lancaster, M. A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol., 10.1016/j.ydbio.2016.06.037 (2016).
- Wevers, N. R. & de Vries, H. E. Morphogens and blood-brain barrier function in health and disease. Tissue barriers 4, 10.1080/21688370.2015.1090524 (2016).
- Schork, N. J. Personalized medicine: Time for one-person trials. Nature 520, 609–11 (2015).
- Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–82 (2012).
謝辞
本研究は、NC3Rsが資金提供するNeuratect CRACK-IT Challenge(プロジェクト番号50308–372160)および、欧州連合のHorizon 2020研究・イノベーションプログラムから助成を受けたCoSTREAM、SysMedPD、BIOPOLコンソーシアム(助成契約番号No. 667375、No. 668738、No. 641639)によって部分的に支援されました。著者らは、ライデン大学分子細胞生物学部門の顕微鏡施設へのアクセスを提供してくださったH.J. Tanke教授、画像取得を支援してくださったJ.C.A.G. Wiegant氏およびA.M.A. van der Laan氏に感謝します。また、神経毒性および電気生理学に関する助言をいただいたR.H.S. Westerink氏とそのチームに感謝します。さらに、技術サポートを提供してくださったモレキュラーデバイスのBen Haworth氏、カルシウムイメージングに関する有益なコメントをいただいたライデン大学医療センター分子細胞生物学部門のS.H. Michel氏に感謝します。
著者の貢献
N.R.W.、R.V.、K.J.W.、C.C.、H.L.は実験の計画または実施を行いました。A.N.とS.J.T.はカルシウムイメージングデータの定量化を設定しました。N.R.W.、R.V.、P.V.はすべての著者の意見を取り入れて本論文を執筆しました。P.V.とJ.J.は研究のすべての側面を監督しました。
追加情報
補足情報は以下のURLで公開されています:https://www.nature.com/srep
競合する財務的利益:すべての著者は、OrganoPlate®を販売するオランダのMIMETAS BVの従業員です。P.V.、J.J.、S.J.T.は同社の株主です。
引用方法:Wevers, N. R. ほか. High-throughput compound evaluation on 3D networks of neurons and glia in a microfluidic platform. Sci. Rep. 6, 38856; doi: 10.1038/srep38856 (2016).
出版社の注記:Springer Natureは、掲載地図や所属機関に関する管轄権の主張について中立の立場を維持します。
本研究は、Creative Commons Attribution 4.0 International Licenseの下でライセンスされています。本論文中の画像やその他の第三者素材は、クレジットラインで別途記載されていない限り、Creative Commonsライセンスに含まれます。ライセンスに含まれない素材を使用する場合は、ライセンス保持者から許可を得る必要があります。このライセンスのコピーは以下で閲覧できます:https://creativecommons.org/licenses/by/4.0/
PDF版(英語)