Application Note 配列特異的DNAアッセイ
PDF版(英語)
はじめに
Threshold® システムを用いて、特定の DNA 配列を測定することができます。測定する標的配列に特異的なビオチン化および蛍光化オリゴヌクレオチドプローブは、Immuno Ligand Assay(ILA)キットのコンポーネントとともに使用されます。プローブは、ターゲットの同じ鎖に、互いに隣接してアニールするように選択されます。測定するDNAサンプルを適切な制限酵素で消化し、小さなDNA断片上の標的配列を遊離させます。消化されたDNAは、過剰のビオチン化プローブと蛍光プローブの存在下で変性されます。プローブと変性した標的はアニールさせられ、プローブ-標的ハイブリッドを生成します。これらのプローブ-ターゲットハイブリッドは、ビオチン化プローブに結合するILA捕捉試薬(ストレプトアビジン)を用いて、ビオチン化Thresholdスティック上に捕捉されます。捕捉されたプローブとターゲットのハイブリッドは、蛍光プローブに結合するILA酵素試薬(抗フルオレセイン-ウレアーゼ結合体)を用いて検出され、定量されます。
配列特異的アッセイには2つのオリゴヌクレオチドプローブが必要で、1つはビオチン化され、もう1つは蛍光化されています。これらは約30塩基の長さで、両方とも標的分子の同じ鎖にハイブリダイズしなければなりません。標的部位を含むDNA断片が大きいと、一般にシグナルが低くなるので、最大の感度を得るためには、サンプルまたは標準DNAを制限酵素で消化して、両方のプローブの結合部位を含む小さな断片を作る必要があります。このため、正確な定量を行うためには、標準品と未知試料の両方に同じ制限酵素を使用する必要があります。
本アプリケーションノートでは、このような特異的配列アッセイの予備的プロトコルを提供します。大腸菌プラスミドpGEM3および関連するpMB1ベースのレプリコン(pBR322など)に対するアッセイの開発と特性評価をモデル系として用いています。このアプリケーションノートはガイドとして作成されたものであり、このアッセイのバリデーションを示すものではなく、また必ずしもすべてのプローブとターゲットDNAの組み合わせに最適な性能パラメータを示すものでもありません。
材料
- Molecular Devices CorporationのThreshold® System(カタログ番号0200-0500)、1311 Orleans Drive, Sunnyvale, CA 94089、TEL:408-747-1700または800-635-5577。
- Molecular Devices CorporationのImmuno-Ligand Assay Detection Kit(カタログ番号R9003)。
- New England Biolabs(カタログ番号139S)の制限酵素HhaI、付属の10X制限バッファーおよび100X BSAとともに使用。
- ビオチン化および蛍光化オリゴヌクレオチドプローブはApplied Biosystemsから購入しました。プローブは、合成中にこれらの位置にビオチンまたはフルオレセイン誘導体化ヌクレオチドを付加することにより、5'末端にビオチン化またはフルオレセイン化しました。使用したプローブは、HhaI制限部位に挟まれたプラスミドの複製起点(ORI)領域の114bpの保存領域に相補的です。プローブの配列は以下の通りである(5'から3'):
ビオチン化:
b-CCAGTTACCTTCGGAAAAAG
蛍光標識:
f-TAGCTCTTGATCCGGCAAACAAACCACCGCTGGTAGCGT - QiagenカラムはQiagen Inc.から購入した(Qiagen-tip 500、カタログ番号10063)。カラムを通すためのバッファー は Qiagen Plasmid Handbook に従って作成しました。
- BSA, fraction VはSigma Chemical Co.から購入し(カタログ番号A7888)、滅菌水に25 mg/mLで溶解して注入しました。
- ハイブリダイゼーションバッファー。ハイブリダイゼーションバッファーの処方は以下の通りである:
| ストック(1リットル) | 数量 | 最終濃度 |
|---|---|---|
| NaH 2 PO 4 .H 2 O |
20.7g | 150 mM |
| NaCl | 26.3g | 450 mM |
| 0.5 M EDTA | 6 mL | 3 mM |
| Triton-X-100 | 2.5 mL | 0.25% v/v |
| 25 mg/mL BSA | 1 mL | 25 µg/mL |
900mLの脱イオン水に撹拌して溶解し、10M NaOH(約10mL)でpH7.4に調整してから最終容量にします。0.2 µmフィルターでろ過滅菌します。
方法
DNAストックの調製、制限消化、アガロースゲル電気泳動には、標準的な分子生物学的手法を用いることができます。必要な試薬の詳細を含め、これらの基本的な技術を網羅した包括的な実験マニュアルがいくつかあります1,2。一般的に、DNAを取り扱う際には、無菌的技術と滅菌されたディスポーザブルを使用すべきです。
ストックDNAの調製
pGEM3プラスミドストックは、Qiagen Plasmid Handbookの指示に従い、Qiagenカラムを用いて調製しました。このストックの濃度は、分光光度計で 260 nm の吸光度を測定して決定しました。50μg/mLのDNAの260 nMにおける吸光度は1 O.D.です。このストックDNAを制限酵素HhaIで37℃で一晩、消化しました。消化の効率はアガロースゲル電気泳動で確認し、フェノール抽出とエタノール沈殿で精製しました。
得られた核酸ペレットをTE(10mM Tris HCl、1mM EDTA pH7.6)に懸濁し、分光光度計で濃度を測定しました。その後、消化DNAをハイブリダイゼーションバッファーで100ng/μLに希釈し、-20℃で保存しました。このストック消化DNAは、標準曲線の作成およびスパイク回収実験におけるプラスミドスパイクに使用しました。
アッセイプロトコール
すべてのハイブリダイゼーションはハイブリダイゼーションバッファー中で行い、検出はThreshold Systemと標準ILA試薬を用いて以下のように行った:
ステップ1 標的DNA(ハイブリダイゼーションバッファー中の標準および消化サンプル+/-スパイク)100 µLを各2 mLサルステットチューブに分注しました。
ステップ2 各チューブに100 µLのプローブミックスを加えます(ハイブリダイゼーションバッファー中の各プローブの濃度は2.4 nM)。短時間ボルテックスして混合します。
ステップ3 チューブを105℃のヒーティングブロックで10分間インキュベートします(変性ステップ)。
ステップ4 すべてのチューブを55℃の加熱ブロックに移し、10分間インキュベートします(アニーリングステップ)。
ステップ5 すべてのチューブを氷に移し、5分間インキュベートします。
ステップ 6 再構成した ILA Capture 試薬 1 mL を ILA Assay Buffer で 1:10 に希釈したものを加えます。チューブにキャップをし、数回転倒させてキャプチャー試薬を混ぜます。
ステップ 7 チューブ全量をろ過ユニットに移し、全サンプルを低真空でスレッショルドスティックにろ過します。
ステップ 8 ILA Wash Buffer 2 mL を各サンプルウェルに加え、高真空でろ過します。
ステップ 9 洗浄液がすべてろ過されたら、SPECIAL バキュームに変更します。酵素試薬を加える前にすべてのウェルが完全に空であることが重要です。そうでないと酵素が希釈され、そ の結果ウェルのシグナルが減少します。
ステップ 10 再構成した ILA 酵素試薬を各ウェルに 100 µL ずつ加えます。メンブレン表面に気泡がないか注意深くチェックします。気泡は酵素のろ過を妨げます。気泡がある場合は、フィルターブロックを手で鋭く叩くと外れます。
ステップ 11 ウェルが空になったら、各サンプルウェルに ILA Wash Buffer 1 mL を加え、高真空でろ過します。
ステップ 12 すべてのウェルが空になったら真空を止め、スティックを読み取ります。
注:ステップ 2、6、8、10、11 の試薬添加は、Combitip® 付きエッペンドルフ・リピータ ー・ピペッターを使用して行うことができます。100µlの添加は、マイクロピペットチップをCombitipに装着するとより正確に行えます。
標準曲線の作成
特定の配列を定量し、スティック間のばらつきを補正するためには、DNA標準曲線を作成し、各スティックの#の位置でミッドキャリブレーターを実行することが有用です。標準品は通常、ハイブリダイゼーションバッファー中の消化pGEM3の2倍希釈系列として、15000 x 106から235 x 106分子/test(45から0.7ng/test)の範囲で実行しました。1875×106分子/testのミッドキャリブレーターとハイブリダイゼーションバッファーのゼロキャリブレーターをそれぞれ#と*の位置で各スティックにかけました。これらの各標準物質100 µLと、ゼロおよびミッドキャリブレーターの複数のアリコート(各スティックに1つずつ)を調製し、アッセイプロトコルに従ってサンプルで処理しました。典型的な標準曲線を図 1 に示します。pGEM3の標準曲線には、一般的にべき乗の式が最も適合することがわかりました。
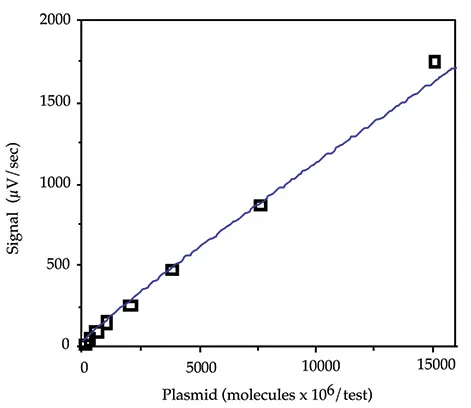
図1: pGEM標準曲線
アッセイ特性
ターゲットフラグメントのサイズ効果
配列特異的アッセイでは、ターゲットフラグメントが小さいほど高い特異的シグナルが得られます。これを実証するために、pGEM3のサンプルをHhaI、HinfI、HindIIIで消化し、それぞれ114、509、2867 bpの断片で標的領域を遊離させました。各消化DNAの20ナノグラムをルーチンのプロトコールに従ってアッセイし、生のシグナルを比較しました。その結果、シグナルを最大にするためには、ターゲット部位はできるだけ小さな断片上で遊離するように選択すべきであることが示唆されました。結果を表1に示します。
| 酵素 | ターゲット長(bp) | シグナル(µV/sec) |
|---|---|---|
| HhaI | 114 | 698.7 |
| HinfI | 509 | 319.5 |
| HindIII | 2867 | 60.5 |
表1:シグナルに対するターゲットサイズの影響
プローブ負荷試験
アッセイに使用するプローブの適切な濃度を決定するために、プローブ負荷試験を行いました。1.2、2.4、6.0、12.0 nM の各プローブを含むプローブミックスを 0、236 x 106、15000 x 106 分子/test の pGEM3 でテストしました。得られたデータを図2に示します。各プローブの濃度が2.4 nMのプローブミックスが選ばれたのは、低バックグラウンド、高シグナル、プローブの経済的な使用という組み合わせが得られたからです。
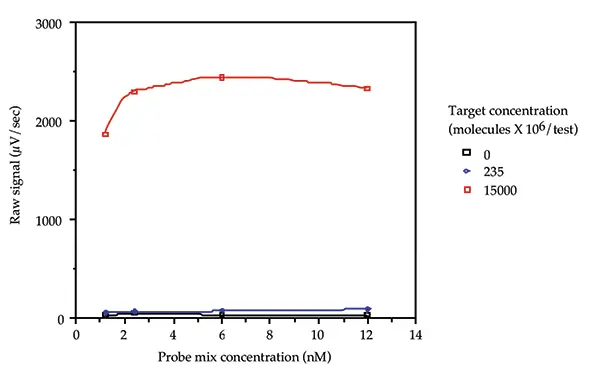
図2:プローブ負荷試験。
変性速度論
二本鎖ターゲットDNAの変性に必要な時間は、高レベルのターゲットDNA(15,000 x 106分子/試験)と0、1、2.5、5、10、15、30、60分の変性時間を用いて決定されました。0分と1分の沸騰ではシグナルが大幅に減少しました。変性時間が1分を超えると、シグナルはかなり高くなりました。ルーチンのプロトコールでは、変性時間として10分を選択しました。
ハイブリダイゼーション速度論
オリゴヌクレオチドプローブが一本鎖ターゲットDNAにハイブリダイズ(アニーリング)するのに必要な時間を、0、235 x 106および30,000 x 106分子/testのターゲットDNAと、0、5、10、15および30分のアニーリング時間を用いて決定しました。データを図3に示します。ルーチンのプロトコールでは10分が最適なハイブリダイゼーション時間として選択されました。
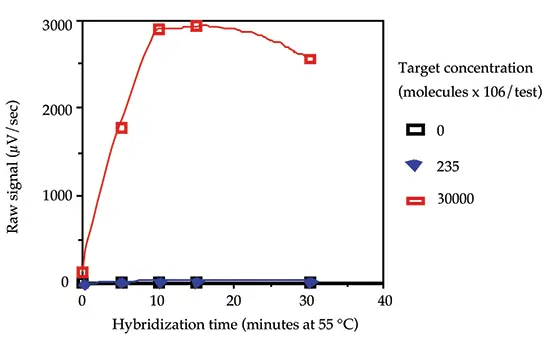
図3:ハイブリダイゼーション速度論。
検出限界
pGEM3プローブアッセイの検出限界は、Threshold System Operator's ManualのILAセクションのガイドラインに従って決定しました。pGEM3アッセイの検出限界は235 x 106 molecules/testで、バックグラウンドシグナルとの標準偏差は4でした。
再現性
アッセイ内再現性は、約 1875 x 106 分子/test の pGEM3 HhaI 消化物の 6 レプリケートをテストすることで評価した。結果を表2に示します:
| 値×10 6 /test) |
|
|---|---|
| 2102.3 | |
| 1889.1 | |
| 1807.3 | |
| 1965.2 | |
| 1856.7 | |
| 平均 | 1915.4 |
| 標準偏差 | 105.0 |
| C.V. | 5.5% |
表2:アッセイ内再現性。
アッセイ間(日間)の再現性は、6つの独立したアッセイを実施することにより、3つの異なる方法で評価しました。第一に、HhaI消化pGEM3のストックサンプルを約1875 x 106 molecules/testに希釈し、各アッセイで3倍希釈してテストしました。第二に、インタクトなpGEM3のサンプルを6回HhaIで消化し、希釈してそれぞれの独立したアッセイで測定しました。第三に、大腸菌DH5α(pGEM3)の全DNAを、インタクトなpGEM3スパイクを含むサンプルと含まないサンプル(1回の試験で約1500×106分子)を、6回HhaIで消化し、希釈して、それぞれの独立したアッセイで測定しました。このようにして、日毎のアッセイの変動性を、日毎の消化による変動性の有無にかかわらず評価することができました。定量結果を表3に示します。大腸菌 DNA 消化物のスパイク回収率は 98%から 128%でした。
| バルク pGEM3 消化物 | 日間 pGEM3 消化物 | 日間 大腸菌 消化物 | |
|---|---|---|---|
| 平均 | 1905.7 | 1550.4 | 2224.8 |
| 標準偏差 | 81.4 | 49.2 | 215.6 |
| c.v. | 4.3% | 3.2% | 9.7% |
表3:日々の再現性(定量×106分子/試験)
特異性
pGEM3アッセイの特異性を調べるため、0、0.1 µg/test、1 µg/testの消化子牛胸腺DNA(HaeIIIで消化した子牛胸腺DNAとRsaIで消化した子牛胸腺DNAの混合物)の存在下で、3段階のpGEM3(0、235 x 106、15000 x 106分子/test)を標準プロトコールに従ってアッセイしました。235×106分子/testのpGEM3サンプル(690 pg/test)、すなわち1500倍過剰の子牛胸腺DNAを1μg/testの子牛胸腺DNAに添加しても、シグナルに有意な増加は見られなかった。1μg/testの子牛胸腺DNAの存在下では、高レベルのpGEM3(15000×106分子/test)からのシグナルの有意な減少は見られなかったことから、子牛胸腺DNAはいずれのプローブとも結合しないことが示唆された。結果を表4に示します:
| 子牛胸腺 DNA 添加 | |||
|---|---|---|---|
| pGEM3 | 0 | 0.1µg | 1µg |
| 0 | 2.2 | 4.7 | 4.8 |
| 235 | 32.9 | 35.7 | 33.2 |
| 15000 | 1414.3 | 1402.4 | 1483.6 |
表 4:非標的 DNA の影響(シグナル単位:µV/sec)
まとめ
本アプリケーションノートに記載されたデータは、HhaI で消化した pGEM3 モデルシステムとシーケンシャル ILA プロトコルを用いて作成されました。標準的なDNA調製で良好な直線性が見られ、アッセイの再現性は優れていました。この配列特異的アッセイはPCR産物の定量に使用されており(3,4)、プラスミドコピー数の決定など、他の様々なアプリケーションにも有用と考えます。
参考文献
- Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., and Struhl, K. Current Protocols in Molecular Biology, Volumes 1 to 3, WileyInterscience (1989).
- Sambrook, J., Fritsch, E.F., Maniatis, T. Molecular Cloning, A Laboratory Manual, 2nd Edition, Volumes 1 to 3, Cold Spring Harbor (1989).
- Olson, J.D., Panfili, P.R., Zuk, R.F., and Sheldon, E.L. シリコンセンサーベースのシステムにおけるDNAハイブリダイゼーションの定量:PCRへの応用。Molecular and Cellular Probes, 5:351-358 (1991).
- PCRと新しいデュアルプローブハイブリダイゼーションフォーマットによるカプセル形成炭疽菌芽胞の同定。Applied and Environmental Microbiology, 60:1622-1625 (1994).
PDF版(英語)