Application Note マイクロプレートリーダーを用いたCRISPR編集細胞の検証:
イメージングとウェスタンブロット検出によるアプローチ
- GFP陽性細胞と非導入細胞を正確に識別することによりトランスフェクション効率を測定
- タンパク質バンドのウェスタンブロット分析を用いてCRISPRを介した遺伝子ノックダウンを検証
- 高品質のウェスタンブロット画像を取得
PDF版(英語)
はじめに
ゲノム編集は、遺伝子発現やタンパク質機能の研究に広く用いられていますが、これらの方法の多くは手間がかかり、精度も低いです *1 。CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9システムは、その精度の高さと使いやすさから、遺伝子編集のためのツールとして非常に普及しています *2。Cas9システムは、細菌や古細菌の自然免疫システムの一部として誕生し、外来DNAから身を守るために用いられてきました *3,*4。既知の外来DNAの短い配列は、CRISPRターゲティングRNA(crRNA)として発現され、Cas9酵素が同様の配列を含む外来DNAを切断するようガイドします。このシステムを利用することで、研究者は遺伝子サイレンシングと転写制御の効果を研究することができ、疾患細胞における遺伝的障害を緩和できる可能性があります *5。
crRNAに似たガイドRNA(gRNA)は、遺伝子内のある領域を標的として設計され、Cas9酵素は宿主細胞のゲノムのこの特異性領域に二本鎖切断を作り出すことができます(図1)。二本鎖切断が生じると、細胞は非相同末端結合(NHEJ)経路または相同組換え(HDR)経路という2つの修復経路のいずれかを経ます。NHEJ経路は一般に、塩基の挿入や欠失(indel)によって遺伝子を破壊するのに使われ、一方、HDR経路は、2つの類似または同一分子のDNA間で配列を交換することによって、レポーター遺伝子や編集配列をノックインするのに使われます *6。
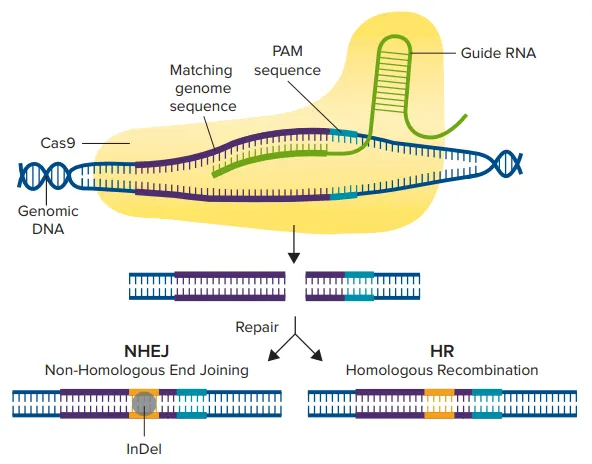
図1. CRISPR/Cas9のメカニズム。Cas9酵素は、まずガイドRNAに結合し、次に3塩基PAM配列の直前にある一致するゲノム配列に結合することで活性化されます。その後、Cas9酵素が二本鎖切断を作り、NHEJまたはHDR経路のいずれかがDNAの修復に使用され、編集された遺伝子配列が得られます。
遺伝子編集のバリデーションは、導入された変異が目的の遺伝子をノックダウンしたかどうかを判定するために必要です。通常、欠失や挿入の成功を確認するためにゲノムPCRや遺伝子シーケンシング検査が行われますが、インフレーム欠失/変異からのシグナルを避けるためには、タンパク質発現を測定することが遺伝子ノックダウンのより明確な指標となります *7。
ここでは、SpectraMax i3x マルチモードマイクロプレートリーダーとScanLater™ウエスタンブロット検出システムを用いて、CRISPR/Cas9ゲノム編集を検証する方法を示します。CRISPRキットを介したOrigeneのATG5ヒト遺伝子ノックアウトキットを用いて、HEK293セルにおけるオートファジー関連5(ATG5)タンパク質の発現をノックダウンし、緑色蛍光タンパク質(GFP)とピューロマイシン耐性遺伝子をノックインしました。ノックダウンはScanLaterウェスタンブロット解析で確認しました。
材料
- ATG5 Human CRISPR Knockout kit via CRISPR (Origene)
- FUGENE HDトランスフェクション試薬 (Promega)
- Opti-MEM 還元血清培地 (ThermoFisher)
- ATG5 に対するウサギポリクローナル抗体 (Origene)
- ATG5 HEK293T細胞一過性過剰発現溶解液 (Origene)
- pCas-Guide-EF1a-GFP プラスミド (Origene cat)
- HEK293細胞(ATCC cat.)
- SpectraMax® i3x マルチモードマイクロプレートリーダー (モレキュラーデバイス cat. #i3x)
◦ScanLater™ウェスタンブロット検出システム(モレキュラーデバイス)
- SpectraMax® MiniMax™ 300イメージングサイトメーター(モレキュラーデバイス)
- ScanLater: Eu-ストレプトアビジン評価キット(モレキュラーデバイス)
- ScanLater: 抗マウス評価キット(モレキュラーデバイス)
- Pierce BCAタンパク質アッセイキット (Thermofisher)
- ピューロマイシン(InvivoGen)
- TGX 12% SDS-PAGEゲル (Bio-Rad)
- TransBlot Turbo Mini-size LF PVDFメンブレン (Bio-Rad)
- Trans-Blot® Turbo™システム (Bio-Rad)
方法
細胞トランスフェクション
HEK293細胞をCostar組織培養処理6-ウェルプレートに1ウェル当たり30万細胞でプレーティングしました。細胞は(1)ガイドベクター(AAGATGTGCTTCGAGATGTG)と(2)ピューロマイシン耐性とGFPの配列を含むドナーベクターでトランスフェクトしました。並行して、3ウェルの細胞を、トランスフェクションコントロールとしてGFPを一過性に発現するpCas-Guide-EF1a-GFPベクターでトランスフェクトしました。6μLのFUGENE HDトランスフェクション試薬と2μgの全DNA(3:1の比率)を用いて細胞をトランスフェクトしました。その後、細胞を37℃で一晩インキュベートした。トランスフェクトされた細胞はGFPを発現し、MiniMaxサイトメーターの緑色蛍光チャンネルで同定・計数されました。総細胞数はStainFree解析を用いて算出し、トランスフェクション効率はSoftMax Proソフトウェアを用いてGFP陽性細胞数を細胞総数で割ることにより測定しました。
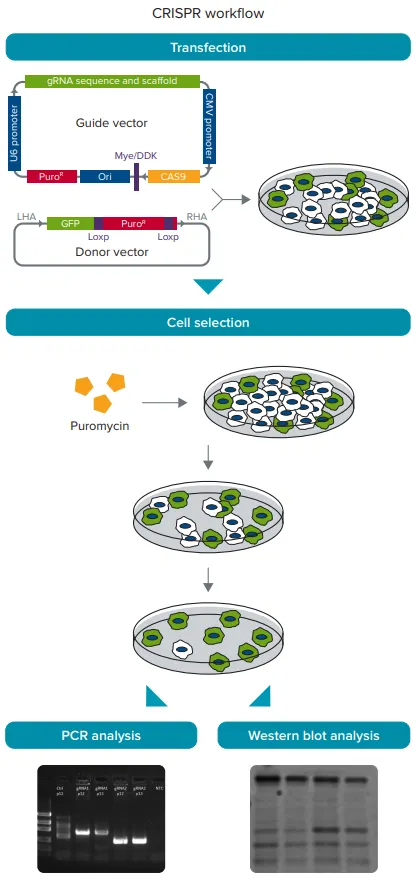
図2. CRISPR/Cas9実験ワークフロー。HEK293細胞にガイドベクターとドナーベクターをトランスフェクト。次に、遺伝子が挿入された細胞を濃縮するために、細胞を1.0μg/mLのピューロマイシンで選択します。選択した細胞からDNAとタンパク質を回収し、ゲノムPCRとウエスタンブロッティングを行い、それぞれ遺伝子の挿入とノックダウンを確認します。
細胞選択
トランスフェクトした細胞を1.0 µg/mLのピューロマイシンを含む培地で培養し、挿入されたピューロマイシン耐性遺伝子を持つ細胞を選択しました。その後、細胞を溶解し、ゲノムDNAと総タンパク質を回収しました。ゲノムDNAはPCR解析のためにGENEWIZに送られ、遺伝子が正しく挿入されていることを確認しました。タンパク質濃度は、SpectraMax i3xリーダーで読み取ったPierce BCAアッセイを用いて定量し、結果はSoftMax Proソフトウェアで事前に設定したプロトコールを用いて解析しました。
ウェスタンブロット
細胞溶解液をLaemmliサンプル緩衝液で希釈し、100℃で10分間煮沸することにより、CRISPR編集細胞と非編集細胞の両方から5μgと10μgの全タンパク質を調製しました。サンプルを4-20%プレキャストゲルにローディングし、SDSPAGEを行いました。その後、Trans-Blot Turboシステムのセミドライ法を用いてタンパク質をPVDFメンブレンに転写しました。
メンブレンはScanLaterブロッキング緩衝液中、室温で1時間インキュベートしました。その後、メンブレンのローディング対照、ラダー、ATG5領域のレーンを別々に切り分け、それぞれの一次抗体(表1参照)中で4℃、一晩インキュベートしました。ScanLater洗浄緩衝液で3回洗浄した後、ブロットをScanLater Eu標識二次抗体と室温で1時間インキュベートしました。抗体とその希釈倍率を表1に示します。
| 一次抗体 | 二次抗体 | |
|---|---|---|
| サンプルブロット | 1:2500 rabbit anti-ATG5 | 1:10000 Goat Anti-Rabbit |
| ローディング対照 | 1:10000 rabbit anti-Vinculin | 1:10000 Goat Anti-Rabbit |
| ScanLaterウェスタンブロットタンパク質ラダー | 1:10000 Eu-Streptavidin |
表1. ウェスタンブロッティングに使用した抗体の希釈率。
二次抗体インキュベーション後、メンブレンをScanLater洗浄緩衝液で3回洗浄し、乾燥させ、ScanLater™ウェスタンブロット検出システムで読み取る前に再度組み立てました。
データ分析
データはSoftMax® ProソフトウェアからImageJソフトウェアにエクスポートされ、相対的なATG5タンパク質発現を定量するためにバンド密度が測定されました。細胞-細胞および細胞-マトリックス接合部に関連する117kDaの細胞骨格タンパク質であるビンキュリンを、サンプルローディングのばらつきを正規化するためのローディング対照として使用しました。
結果
MiniMaxイメージングサイトメーターはトランスフェクトした細胞とトランスフェクトしていない細胞の両方の高画質画像を撮影し、SoftMax® ProソフトウェアはGFP陽性細胞の数と細胞の総数を同定し、定量することができました。トランスフェクション効率は13%と算出されました(図3)。
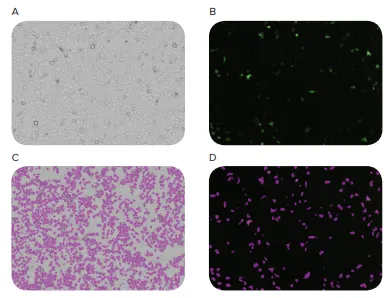
図3. 細胞トランスフェクションの画像。HEK293細胞をpCas-Guide-EF1a-GFPベクターでトランスフェクトし、CRISPR/Cas9トランスフェクト細胞と並行してトランスフェクション効率を計算しました。MiniMaxサイトメーターを用いて、明視野チャンネル(A)と緑色蛍光チャンネル(B)で細胞をイメージングしました。SoftMax Proソフトウェアで両チャンネルの細胞を同定・計数し(CとD)、GFP陽性細胞を全細胞数で割って13%のトランスフェクション効率を算出しました。
ゲノムDNAサンプルのPCR解析から、HEK293細胞ゲノムのATG5領域に正しいサイズの挿入がなされたことが判明しました(データは示さず)。
ウェスタンブロット画像では、CRISPR編集細胞と非編集細胞(陰性対照)との間で、ATG5タンパク質バンド強度の目に見える減少が見られました(図4)。ImageJソフトウェアを用いてタンパク質のバンド密度を計算すると、CRISPR編集細胞は未編集細胞と比較して、ATG5タンパク質の発現がおよそ60%ノックダウンしていました(図5)。
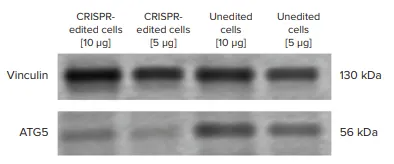
図4. ATG5のウェスタンブロット実験。ATG5タンパク質の発現は、編集していない細胞に比べてCRISPRした細胞では目に見えて低かったです。サンプルローディングの過不足を考慮し、5μgと10μgのサンプルをSDS-PAGEゲルにロードしました。サンプルローディングのばらつきを正規化するため、ローディング対照としてVinculinを用いました。ウェスタンブロットの画像をImageJソフトウェアにエクスポートし、バンド密度を測定しました。
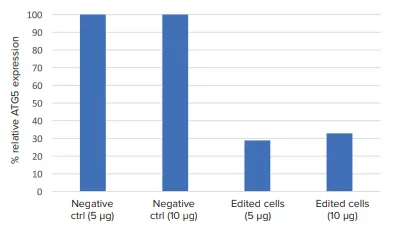
図5. 相対的ATG5タンパク質発現。図4のウエスタンブロット画像をImageJソフトウェアにエクスポートし、バンド密度を解析しました。ATG5タンパク質のバンド密度は、それぞれのローディング対照に対して正規化しました。CRISPR編集細胞は、非編集細胞と比較してATG5タンパク質発現が一貫して低かったです。
結論
CRISPR遺伝子編集技術では、正確な結果を得るためにプロセス全体を注意深くモニタリングする必要があります。SpectraMax i3x マルチモードマイクロプレートリーダーは、最初のトランスフェクションからタンパク質ノックダウンの確認まで、CRISPR編集実験の結果を分析するための完全なソリューションを提供します。MiniMaxイメージングサイトメーターを使用すると、研究者は、総非標識細胞数と蛍光発現トランスフェクト細胞数を比較することにより、トランスフェクション効率を評価することができます。ScanLaterウエスタンブロット検出システムは、コントロール細胞とCRISPR導入細胞における目的のタンパク質の高感度検出と定量分析を可能にします。
参考文献
- Lombardo, A et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nature Biotechnology 25.11 (2007): 1298.
- Barrangou, R. Cas9 targeting and the CRISPR revolution. Science 344.6185 (2014): 707-708.
- Ishino, Y et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology 169.12 (1987): 5429-5433.
- Barrangou, R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315.5819 (2007): 1709-1712.
- Barrangou, R et al. Applications of CRISPR technologies in research and beyond. Nature Biotechnology 34.9 (2016): 933-941.
- Liang, F et al. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proceedings of the National Academy of Sciences 95.9 (1998): 5172-5177.
- Estep, JA et al. Immunoblot screening of CRISPR/Cas9-mediated gene knockouts without selection. BMC molecular biology 17.1 (2016): 9.
PDF版(英語)